|
筋萎縮性側索硬化症(ALS)について
筋萎縮性側索硬化症 (amyotrophic lateral sclerosis, ALS, Gehrig病)
筋萎縮性側索硬化症は、上位・下位運動ニューロンを系統的におかす変性疾患で、
大脳皮質運動領野Betz細胞,錐体路、下部脳幹の運動性脳神経核及び脊髄前角細
胞の著明な変性脱落を特徴とする。40〜50歳代の発症が多いが、10歳代から80歳代
にわたり、男性にやや多い(男性:女性 1.1〜3.0:1)。
大部分は弧発例で、5〜10%に家族性発症が認められる。平均罹病期間が3〜5年と
予後は一般に不良とされる。有病率は世界の各地域ともほぼ同一で、人口10万人当
たり0.8〜6.4/10万人、年間発病率0.4〜2.6/10万人である。
しかしGuam島と日本の紀伊半島、West New Guineaには、有病率が他地域の10数
倍〜100倍と極めて高い多発地域が認められている。
遺伝性筋萎縮性側索硬化症
ALSの5〜10%に常染色体優性遺伝を示す家族性ALSがあり、病理学的に2型に分
類される。
・孤発性ALSと同様の病理所見をしめすもの。
・後索型ALS:脊髄前角、側索に加え、後索のmiddle root zoneに変性を認める。
後索型家族性ALSの20%に於いて第21染色体長腕上のCu/Zn superoxide
dismutase (SOD 1)に数多くのミスセンス変異が同定された。
Kugelberg-Welander病
常染色体劣性遺伝性疾患で、通常発症年齢は2〜17歳位、中年発症もある。
肢帯型筋ジストロフィー類似の症状即ち近位筋優位の筋力低下、筋萎縮を示すが、
下位運動ニューロンの選択的変性が認められる。
Werdnig-Hoffmann病
乳児発症のKW病と考えられている。floppy infant。第5染色体長腕のKugelberg-
Welander病と同一遺伝子に異なる遺伝子異常の生じたもの。
多発地ALS
孤発性ALSと同様の病理所見に加え、大脳皮質、側頭葉海馬回など広汎に神経原
線維変化が出現する。
Kennedy-Alter-Sung症候群(Bulbospinal muscular atrophy, BSMA)
伴性劣性遺伝で、発症年齢は10〜20歳代、筋萎縮の分布は、舌、構音筋など球筋
と四肢筋位筋がおかされる。他に手指姿勢振戦、女性化乳房、不妊等を伴うことがあ
る。やや進行が遅いが、歩行不能になることもある。血清 CKは軽度上昇。
X 染色体長腕にあるアンドロジェン受容体遺伝子の第1エクソンの一部にCAG
(cytocine,adenine,guanine)の繰り返し配列がある。
対照ではリピート数は21個に対し、発症者では40〜52歳代に増加。
女性保因者では正常と異常のheterozygoteになっている。
ALSの病理
弧発性ALSの病理学的特徴は、大脳皮質運動領野、錐体路、脳神経運動核、脊髄
前角神経細胞の選択的な変性脱落及びgliosisであり、脊髄前角及び脳幹部舌下神経
核などの残存神経細胞胞体内にBunina body、ubiquitin陽性封入体、嗜銀性封入体等
を認める。動眼神経、滑車神経、外転神経核、仙髄Onuf核は一般的に障害されない
が、Bunina body、conglomerate inclusion(neurofilamentの細胞内異常蓄積)、あるい
はubiquitin陽性封入体(Lewy body-like inclusion)が動眼神経核やOnuf核に見られた
例が報告されている(Tomonaga 1980, Kihira et al.1997)。臨床経過の早い症例で、
脊髄前角にspheroid即ち近位軸索内に神経細繊維(neurofilament)が異常に蓄積する
変化が多発し、neurofilamentは神経細胞にも異常蓄積し、chromatolytic changeを
示す。ALS脊髄前角では形態的変化が見られない一見正常に見える細胞でも、組織
計測的には核、核小体のサイズの縮小が認められ、経過とともに細胞体の萎縮が著
明となる(Kihira et al.1998)。
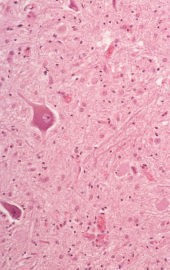 図 : ALS患者の脊髄前角において著しい神経細胞の脱落と 図 : ALS患者の脊髄前角において著しい神経細胞の脱落と
グリオーシスを認める。
多発地ALSの臨床的・病理学的特徴
Guam島,紀伊半島の多発地ALSは一般的には家族内発症が多く、生存期間が他地
域より長く、痴呆などの精神症状を合併する頻度が高い。さらに同じ地域、同じ家系内
にParkinson 痴呆症候群、精神症状を示す例が高頻度にみられることが特徴である。
ただし詳述すると紀伊半島古座川地区と穂原地区ではやや臨床的・病理学的特徴
が異なる。
病理学的には、弧発性ALSと同様上位・下位運動ニューロン障害を認め、これに加
え多発地ALSではAlzheimer 神経原繊維変化(NFT)を海馬のAmmon角、大脳皮質、
大脳基底核、脳幹部に広汎に認める。顆粒空胞変性は多発地ALSの海馬において認
められるが老人斑が認められないことがAlzheimer病とは異なる。NFTの出現の程度は
Guam ALS/PDでもっとも多く、葛原らの穂原地区のKii ALS/PDでも極めて多い。古座
川地区Kii ALSではこれらの例ほど多くはない。
病因
ALSの原因や発病メカニズムはなお不明であるが,仮説としてフリーラジカルによる
細胞障害、superoxide dismutase 遺伝子異常、興奮性アミノ酸であるグルタミン酸によ
る細胞障害、Caイオンの細胞内流入による細胞死、neurofilament異常説、一酸化窒
素(NO)の細胞障害説などが注目されている。多発地ALS ではこれら以外に特に環境
要因、中毒、種特異性などが検討されている。
以上をまとめると筋萎縮性側索硬化症とは
#選択的に運動ニューロンが進行性に障害される原因不明の疾患であり
#上位運動ニューロン(錐体路)、下位運動ニューロン(脊髄前核細胞、脳神経運動核
細胞)がともに侵される。
疫学:人口10万人当たり1.9〜6.5世界各地でもほぼ同様。ただし以下の多発地域が
ある。
1)マリアナ群島グアム
2)紀伊半島南部;古座川町、三重県渡会郡南勢町穂原
3)西ニューギニア、グルートアイランド
歴史
Charcot & Joffroy(1869年)がALSの疾患単位を確立。すなわち臨床・病理学的
観点から脊髄前角と側索の両方が障害されることによって筋萎縮が発症し、経過の早
い症例群を一つの疾患単位として確立。
Duchenne(1860年)Charcot (1870年)が進行性の延髄運動神経核の障害による
構音・嚥下障害を呈する症例群を進行性球麻痺として分類。
Dejerine
(1883年)筋萎縮性側索硬化症と進行性球麻痺が病理学的には同一疾患
であると示した。
臨床症状
1)発症年齢:40〜50歳代が半分を占める。男性:女性 1.1〜3.0 : 1
2)初発症状:上肢遠位部の筋萎縮と筋力低下、下肢の筋萎縮、構音・嚥下障害
3)臨床経過:脊髄前角障害→四肢筋や躯幹筋の萎縮と筋力低下、線維束攣縮
脳神経運動核障害→構音・嚥下障害、舌萎縮、線維束攣縮
錐体路障害→四肢の痙性麻痺、腱反射亢進、病的反射陽性
4)陰性徴候:①感覚障害がない
②眼球運動障害がない
③膀胱直腸障害がない(Onufrowicz核が保たれる)
④褥瘡ができない
検査所見
筋電図:安静時のfibrillation potential
高振幅のNMUでneurogenic pattern
臨床的に正常に見える筋にも広範囲に上記所見が出現する場合、及び
脳神経領域に認められる場合診断価値が高い
筋生検:典型的な神経原性萎縮所見(大小のgroup atrophyや筋束萎縮、
標的線維target fiberの出現、fiber type grouping)
髄 液:蛋白が軽度上昇することも(100mg/dl以下にとどまる)
血清CK:通常は正常、稀に軽度上昇
鑑別診断
1)形性頚椎症、頚椎後縦靭帯硬化症(OPLL):
ALSとの合併例に注意、手術でALS症状の増悪例が多い。
2)頚髄部腫瘍:
通常は上肢の筋萎縮と麻痺、下肢の痙性対麻痺を生じる。しかし通常は
感覚障害を伴い膀胱直腸障害も高率に合併。MRI。
3)頸肋:片側ないし両側の小手筋萎縮と自発痛、感覚鈍麻あり。
4)頭蓋・頚椎移行部の異常、大後頭孔の腫瘍、Arnold-Chiari奇形
5)脊髄空洞症
6)若年性一側上肢筋萎縮症(平山型):
小手筋、前腕の末梢部と近位側の尺側部に限局した筋萎縮
(segmental)で線維束攣縮を伴うが非進行性、10〜20代の男性に多く、首の
激しい運動、外傷歴のあることが多い。頚椎の屈曲位における頚髄圧迫によ
るflexion myelopathyで、診断は頚部MRIで、特にneutral、flexion位などを調
べる必要がある。
7)脳血管障害による仮性球麻痺:
散在性の多発脳梗塞によるもの。構音・嚥下障害著明。舌萎縮がない。
8)多発ニューロパチー:
通常は感覚障害を伴っている。末梢神経刺激伝導速度低下、神経生検所見、
髄液所見、基礎疾患などが鑑別の参考になる。
Multifocal demyelinating motor neuropathy (multifocal motor neuropathy
with conduction block):臨床像はALSに類似。免疫グロブリン大量療法など
治療に反応する。
Chronic inflammatory demyelinating polyneuropathy, CIDP:
通常は感覚運動型。末梢神経刺激伝導速度の低下、神経生検で末梢神経
の脱髄性病変。
Charcot-Marie-Tooth病:常染色体優性遺伝。大腿部下3分の1以下の逆シャン
ペンビン型筋萎縮。感覚障害を伴う。神経伝導速度の低下するもの(1型)と低
下しないもの(2型)があり、1型が多い。
9)多発筋炎:四肢近位筋優位の筋萎縮と筋力低下。深部反射低下。
血清CK値上昇。炎症反応。
10)重症筋無力症:高度の球麻痺が出現するので本症の球麻痺と似るが、眼瞼下垂
と眼球運動障害が高頻度に出現し症状の日内変動がある、テン
シロンテスト陽性などから鑑別。
筋萎縮性側索硬化症の合併症
球麻痺・仮性球麻痺による嚥下障害のため、誤嚥性肺炎を生じやすい。筋力低下や
呼吸筋麻痺のために喀痰排出が困難で気道感染症や窒息が高頻度。一般に慢性神
経疾患に多い褥瘡と尿路感染症は非常に少ない(陰性徴候)。
筋萎縮性側索硬化症の経過と予後
常に進行性であるが、経過は症例毎に異なり、発症から死亡までの期間は、数ヵ月
から10年を越えるものまである。
治療
1)興奮性アミノ酸仮説に基づくリルゾール経口投与
2)呼吸障害に対する治療(鼻マスク、気管切開、人工呼吸器)
3)嚥下障害に対する治療(経鼻栄養、PEG)
4)意志の疎通に関する療法(文字盤、コンピュータなど)
5)適度な運動による筋力保持と関節拘縮の予防
6)その他、患者および家族への病名告知の問題、在宅・施設療養支援
文献
1)豊倉康夫編集:神経内科学書,1996,朝倉書店
2)水野美邦編集:神経内科ハンドブック鑑別診断と治療第2版,1999,
医学書院
|